LEDGER 2.7 ·
503B ·
21 CFR PART 11 ·
cGMP ·
USP <797>/<800> ·
GAMP 5
Operating ERP for sterile pharmaceutical production.
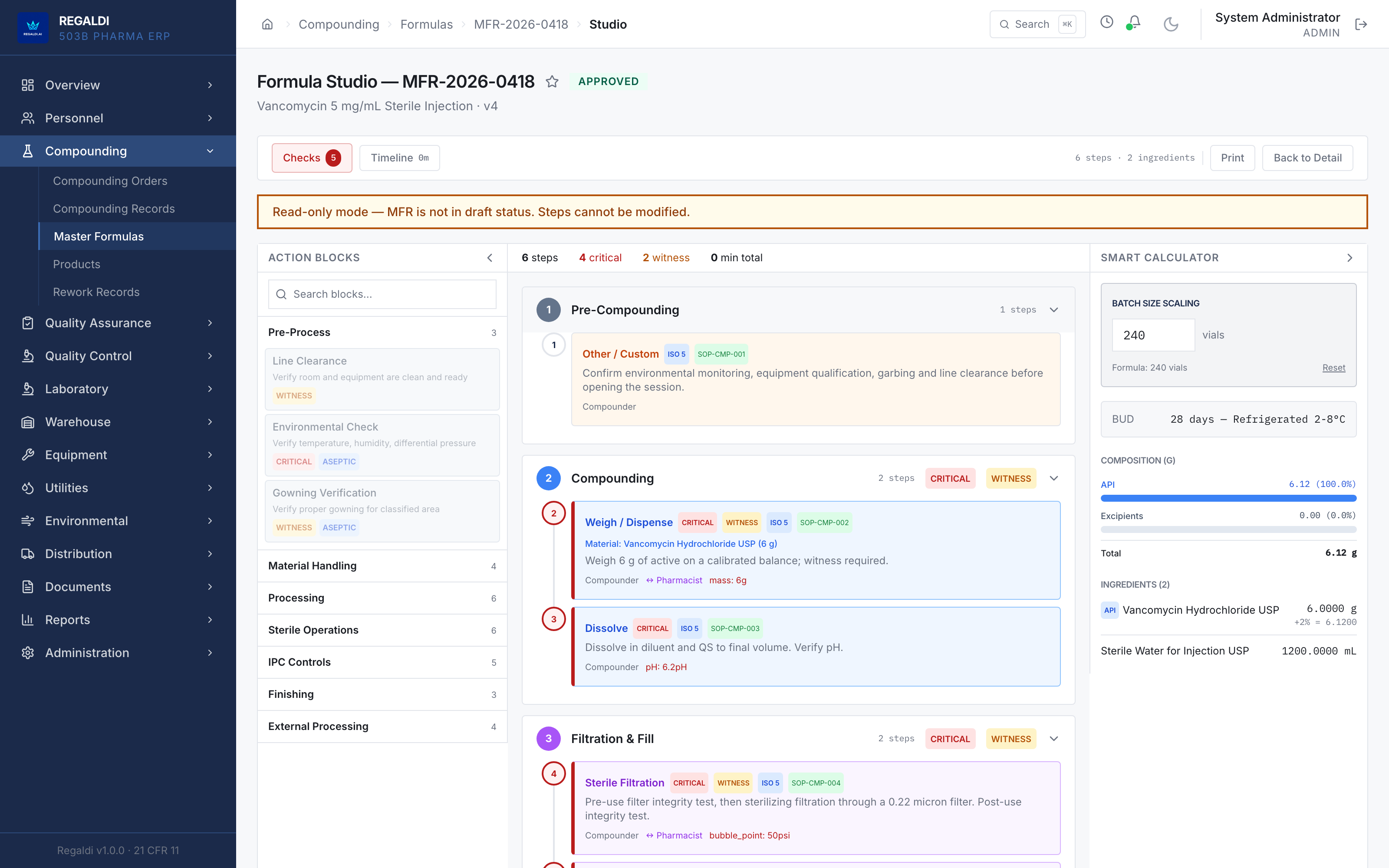
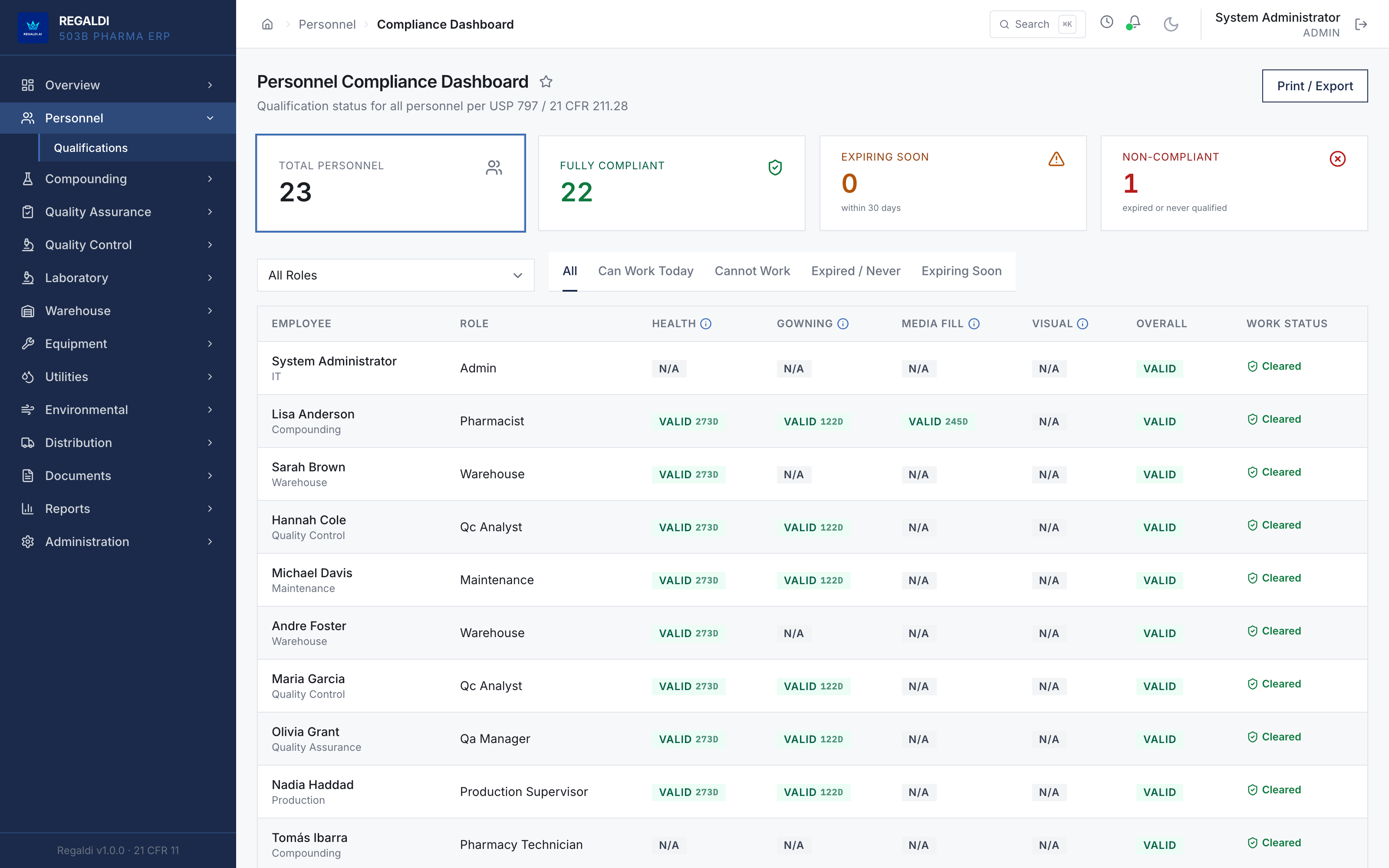
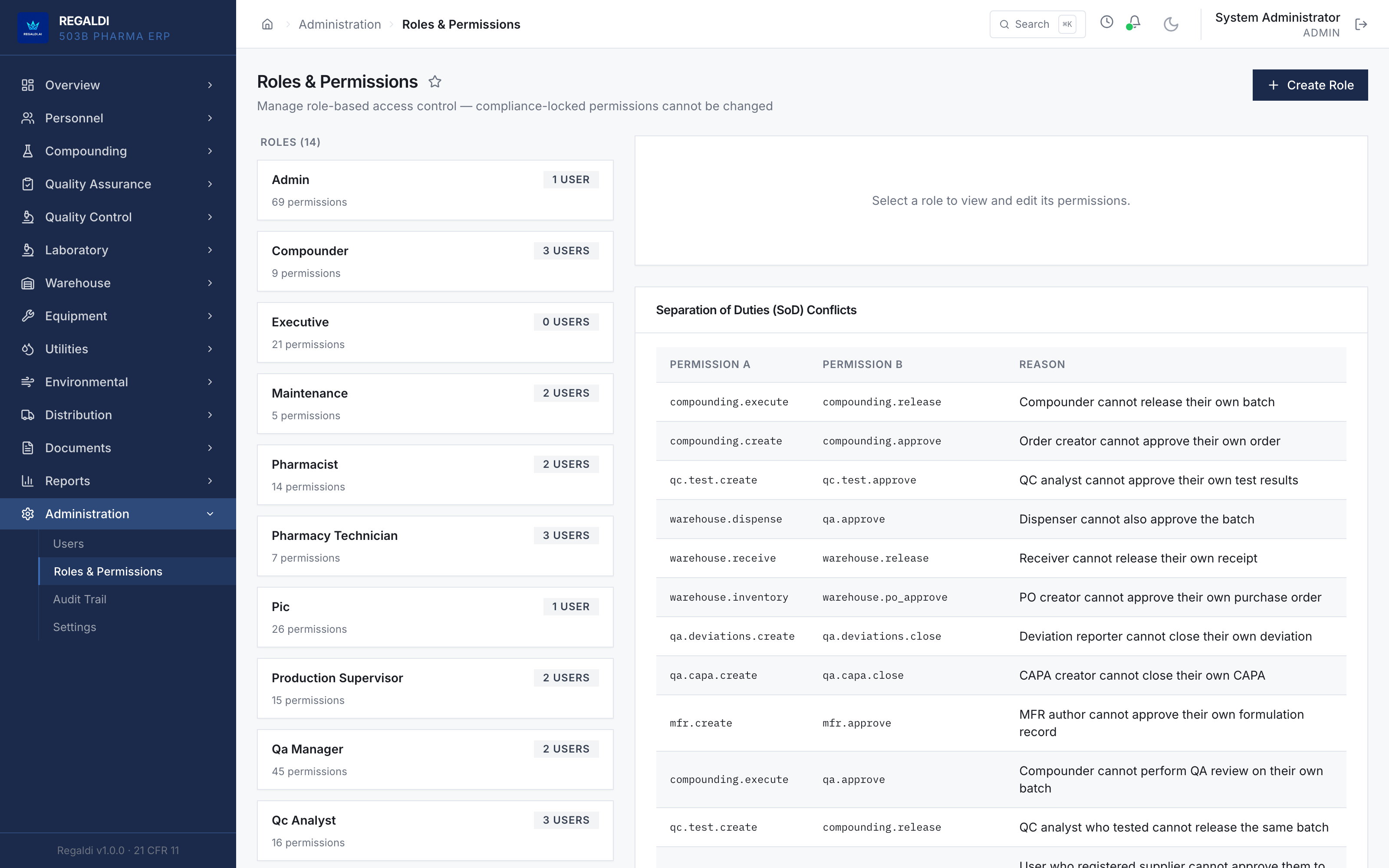
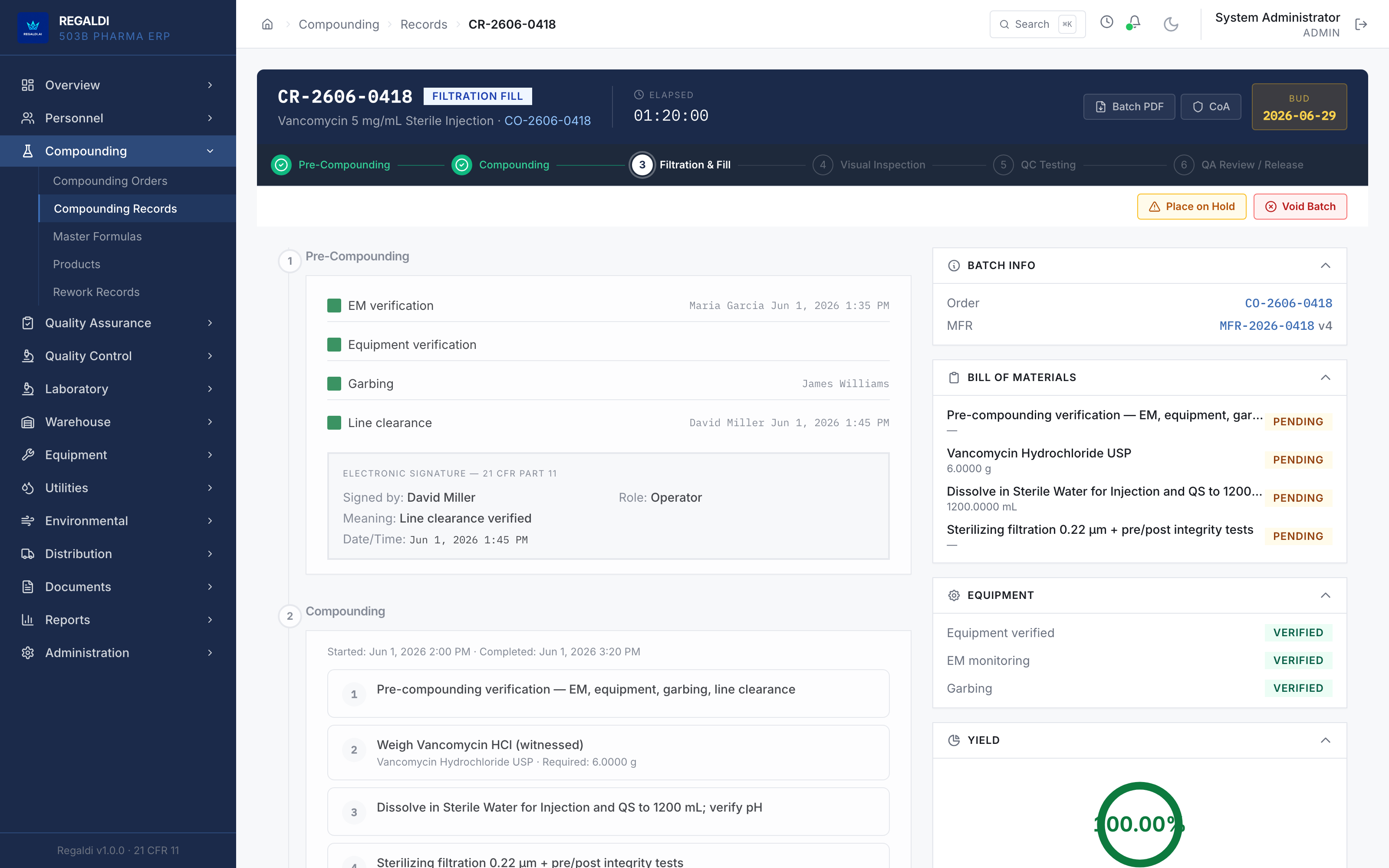
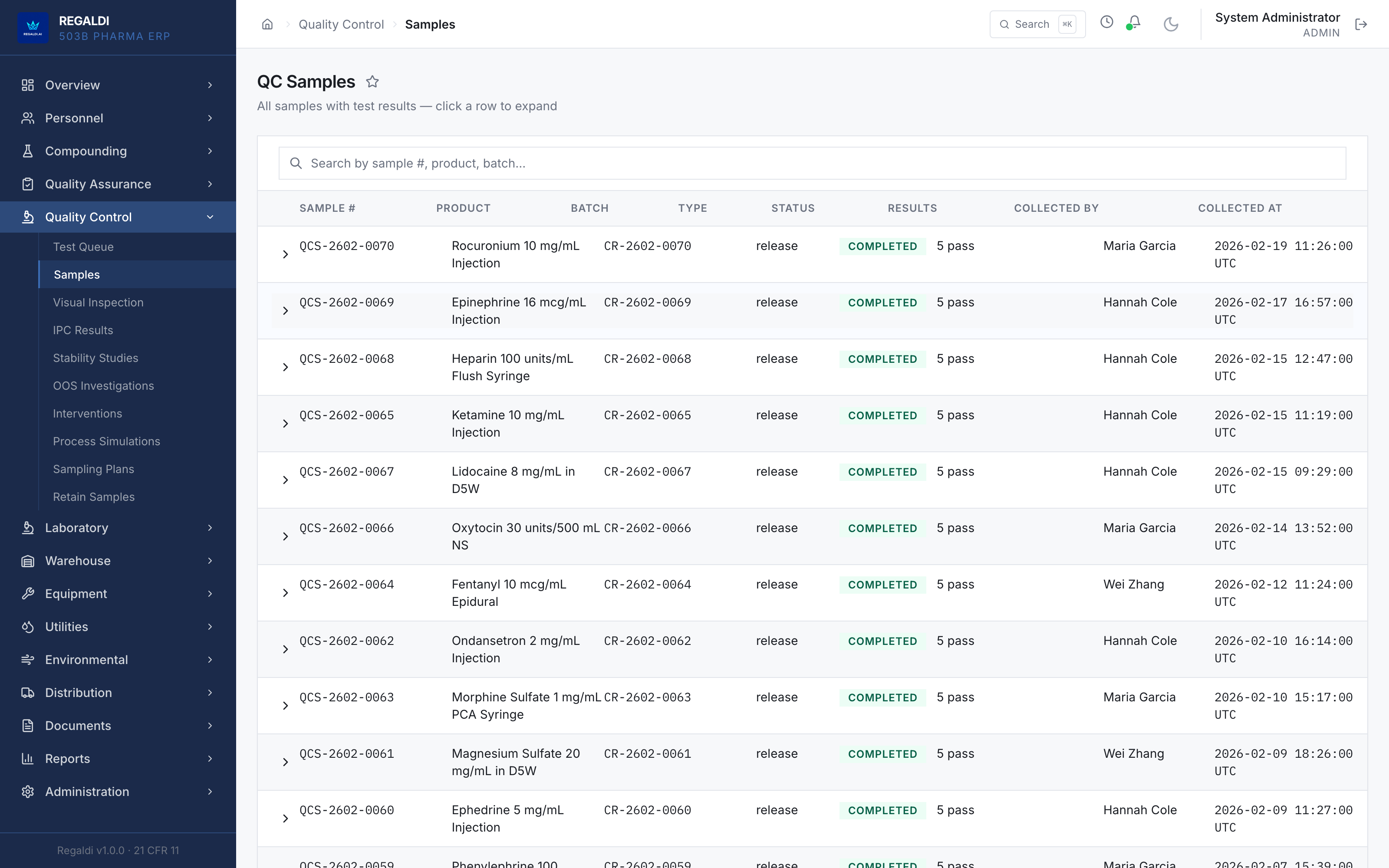
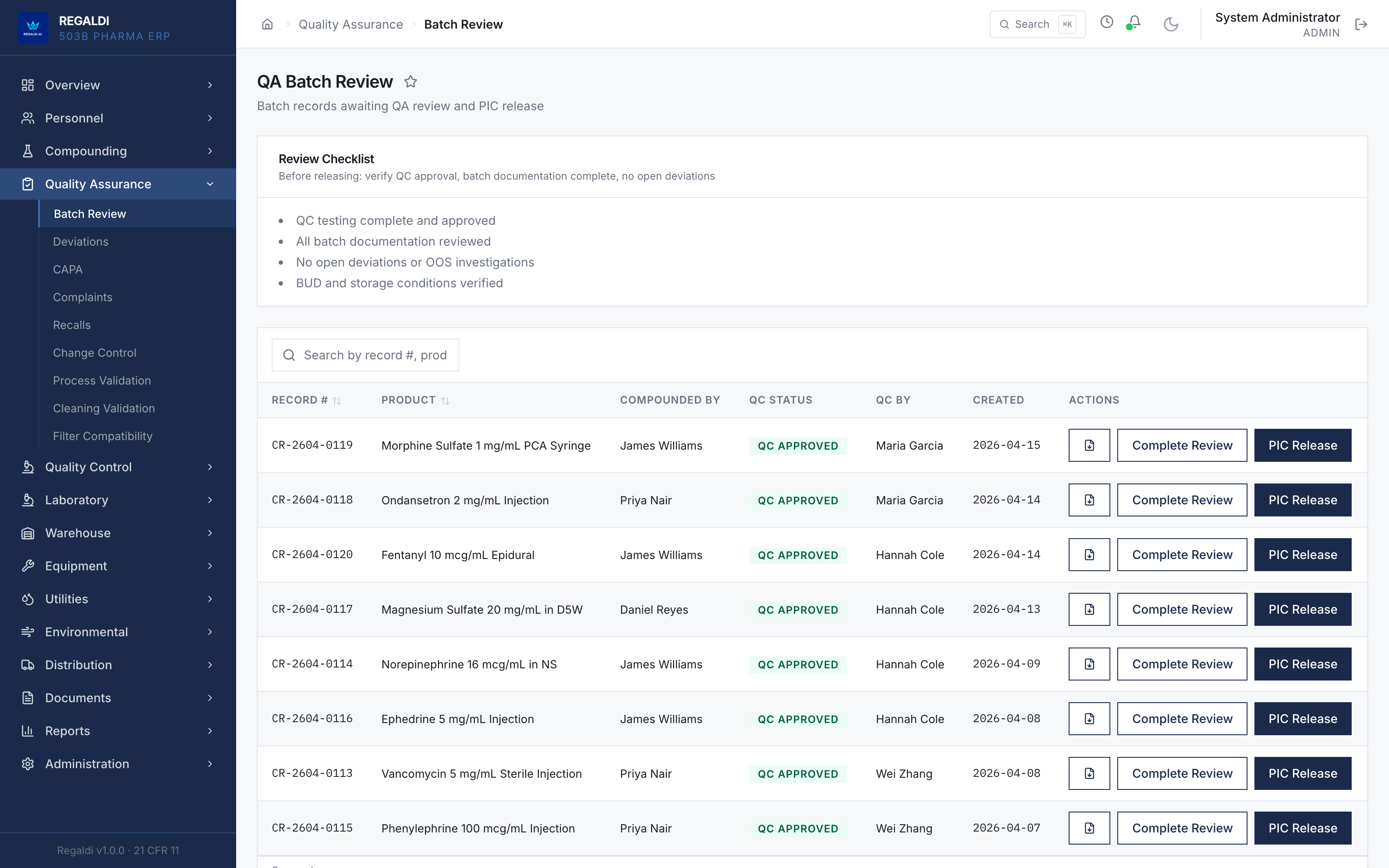
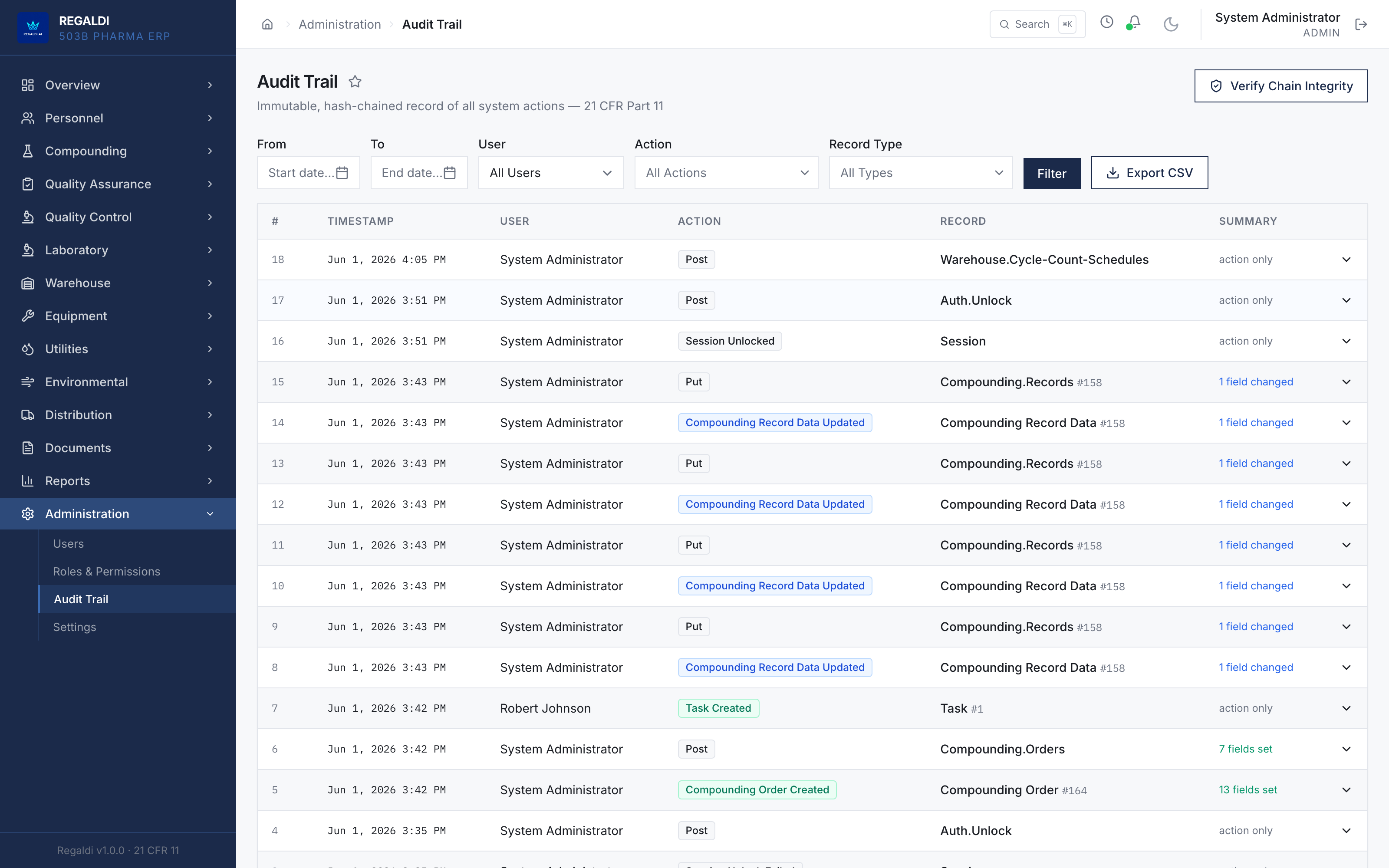
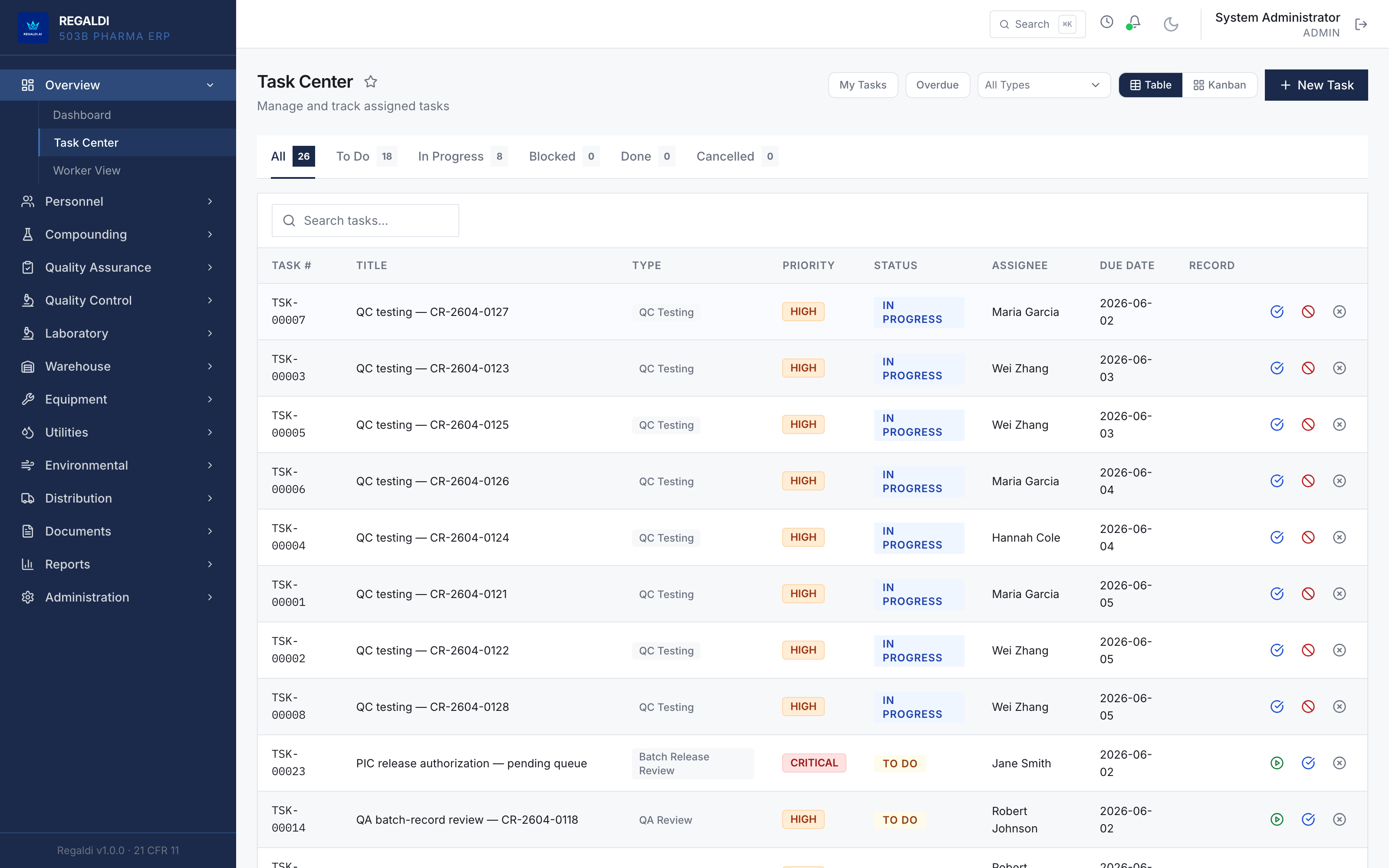
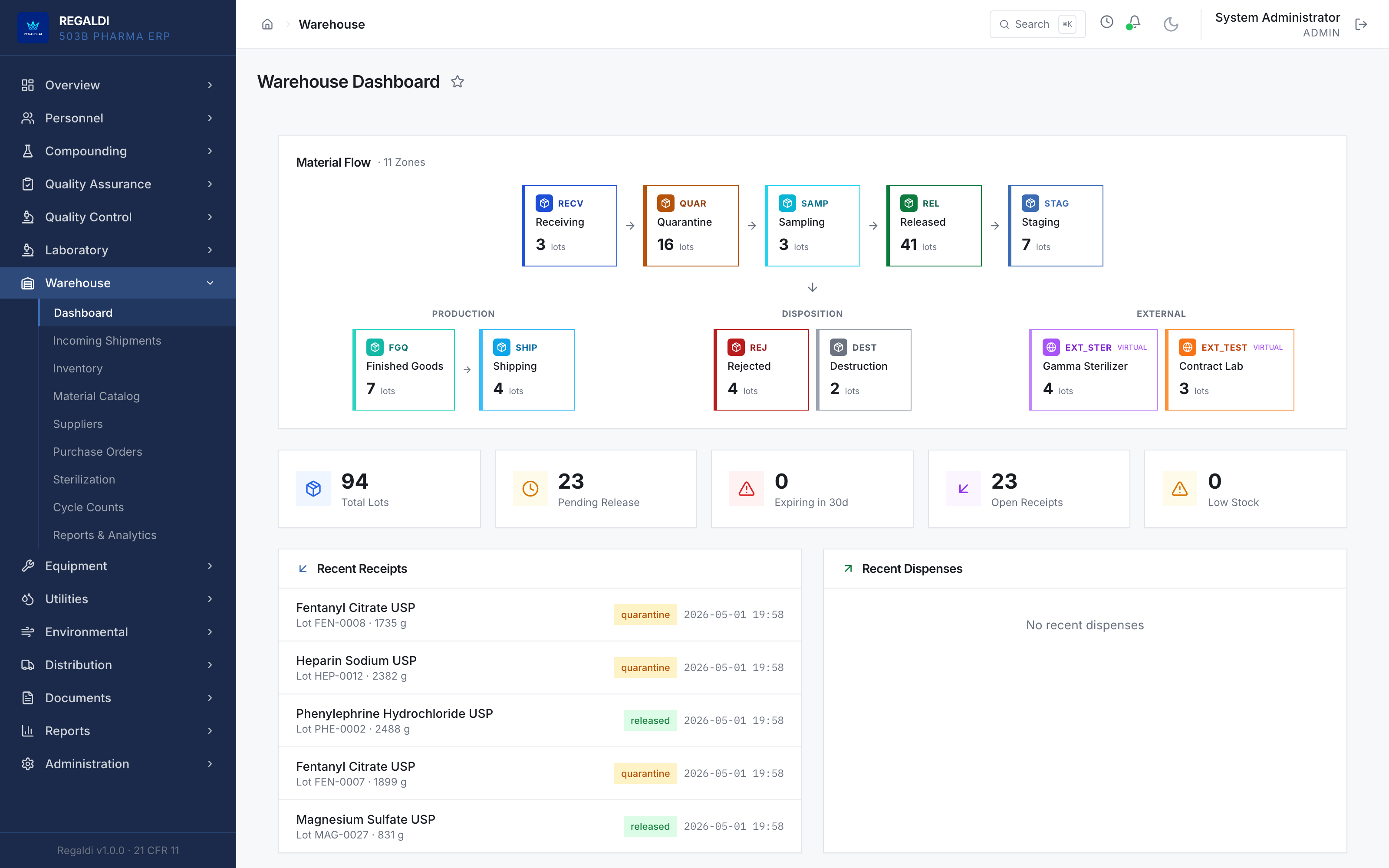
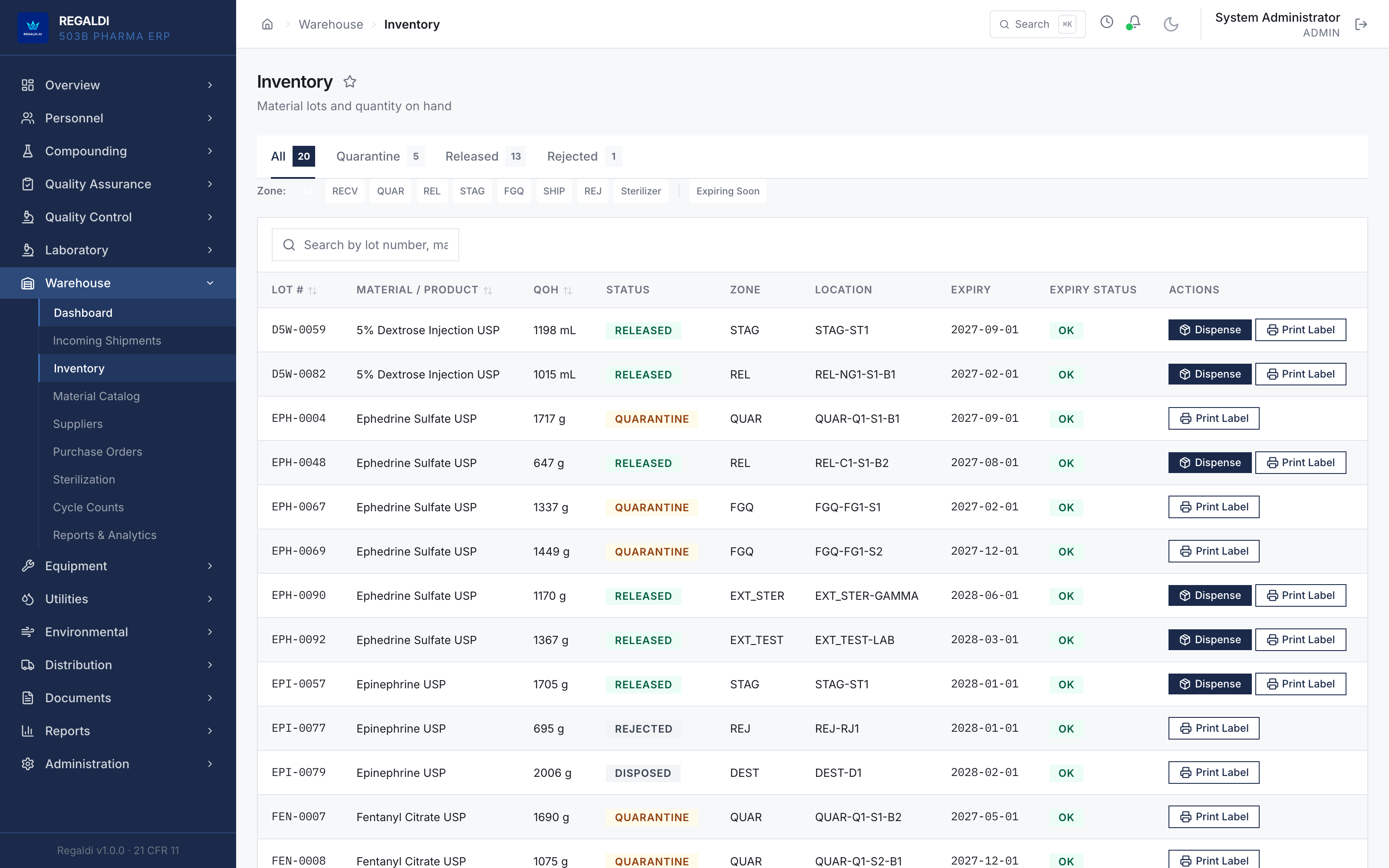
Ledger is the record a sterile-production facility runs on: every batch, every check, every signature, written down as the work happens. Because the governing regulation is recorded beside each action, the production history is inspection-ready as a byproduct of the work, not a reconstruction after it. The whole operation lives in one database: formula, batch, personnel, equipment, environmental monitoring, warehouse, distribution, and documents. A question about any of them is answered by query against one record, and the assistant reads all of it. It is built for FDA-regulated production under current Good Manufacturing Practice (compounding facilities, 503B outsourcing facilities, and contract manufacturers) and implements 21 CFR Part 11 natively from the first commit: Master Formula Studio, the electronic batch record and its six-phase gate, environmental monitoring bound to the compounding session, a SHA-256 hash-chained audit trail, and multi-signature release. The software supports the licensed operators; it does not replace a required signature.
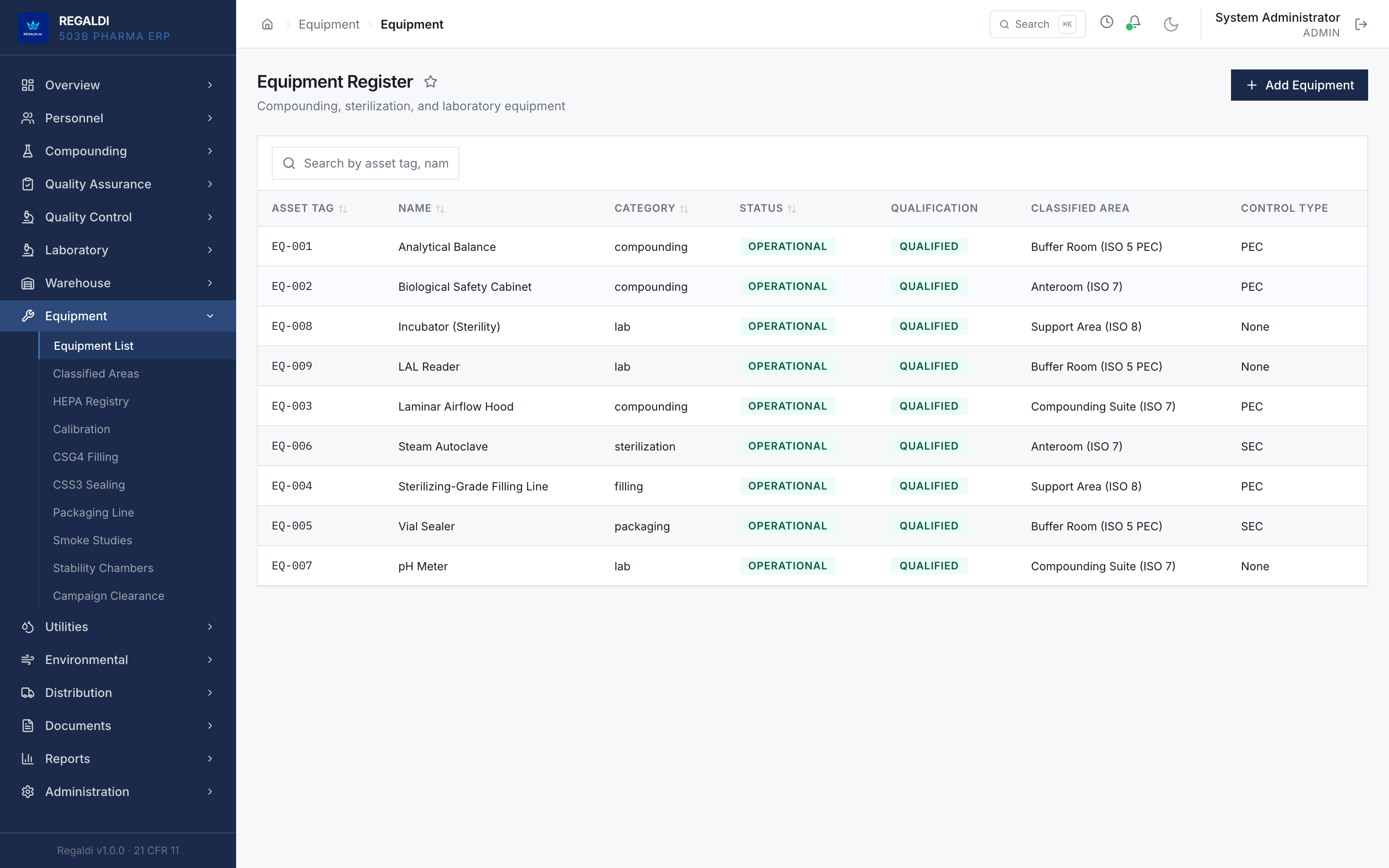
WORKED EXAMPLE · a 503B sterile-compounding outsourcing facility. Ledger is an ERP for 503B outsourcing facilities and sterile-production operations in this regulatory class. The system carries an authored GAMP 5 Category 5 validation package; execution of the package precedes commercial activation. The SCADA and equipment-interface layer is vendor-neutral, with Siemens process equipment as the currently deployed instance. The facility is built; the system awaits commercial activation. Last verified: June 2026.
ABOUT THIS ENGAGEMENT
Regaldi is the software vendor for the facility's system of record. As of this writing the facility is built and awaiting commercial activation; it has not yet produced commercial batches and has no production or inspection history. The capabilities below are functions of the software, which operates on data and configuration supplied by the facility. The software supports the facility's regulatory, quality, sterility, and Board of Pharmacy obligations; it does not replace, perform, or guarantee them. Those obligations remain the facility's responsibility under the supervision of its licensed personnel. Computerized-system validation under GAMP 5 is software validation, not FDA approval, clearance, or inspection.